Background
DNA Barcoding is a technique for identifying species using DNA sequences. Short DNA sequences are used like barcodes on items at the supermarket to uniquely identify each product. Read more about DNA Barcoding on Wikipedia and the website of the Consortium for the Barcode of Life.
In this tutorial we’re going to use a short sequence from the Internal Transcribed Spacer (ITS) which is part of the genes that encode ribosomal RNAs. We’ll use it to identify a plant pathogenic fungus.
Imagine that you are working in a plant disease diagnosis clinic. A strawberry grower has sent you a sample of diseased strawberries. You have isolated a fungus from the infected fruits and identified it as a member of the genus Colletotrichum. Colletotrichum spores all seem to look alike to you and you cannot identify the species based only on spore and colony morphology. You decide to sequence the ITS region so you can read this fungus’ barcode and identify the species.
You use PCR to amplify the ITS region, then you send the PCR product to the sequencing lab. The sequencing lab sends you two chromatograms. Now you need to use the two chromatograms to identify the fungus. We will use Geneious to analyze these sequences.
Procedure
Step 1: Assemble the reads and make a consensus sequence
Download the chromatograms of the two sequence reads: chromatogram 1, chromatogram 2.
These two sequences are from a PCR product that is about 500 bp in length. The sequencing reactions were done with the same primers used to do the PCR reaction, so the two sequences should overlap. We will use Geneious to align the sequences, fix any errors, and prepare a final consensus sequence.
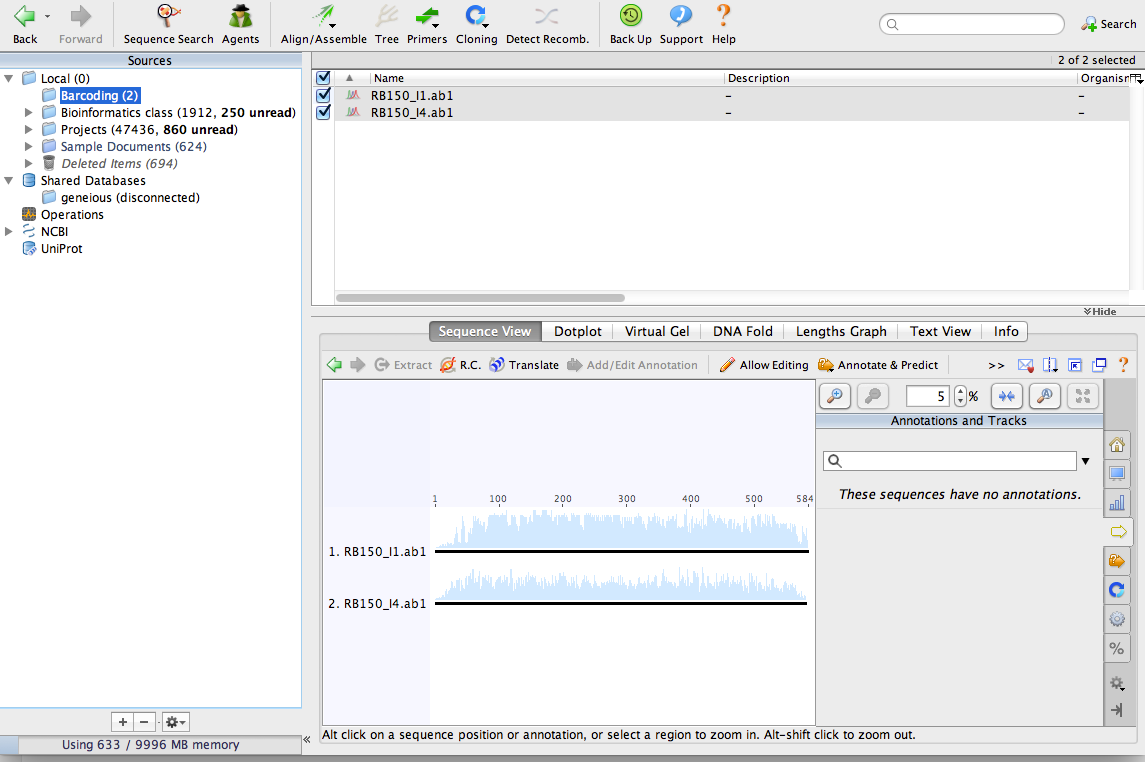
Start Geneious and make a new folder for your barcoding project. From the menu at the top of the screen select File > New Folder.
Import the chromatograms into the new folder. You can just drag them into the Geneious window to import them. After importing the files, your window should look something like this:
Use the zoom buttons to zoom in to see the DNA sequences:
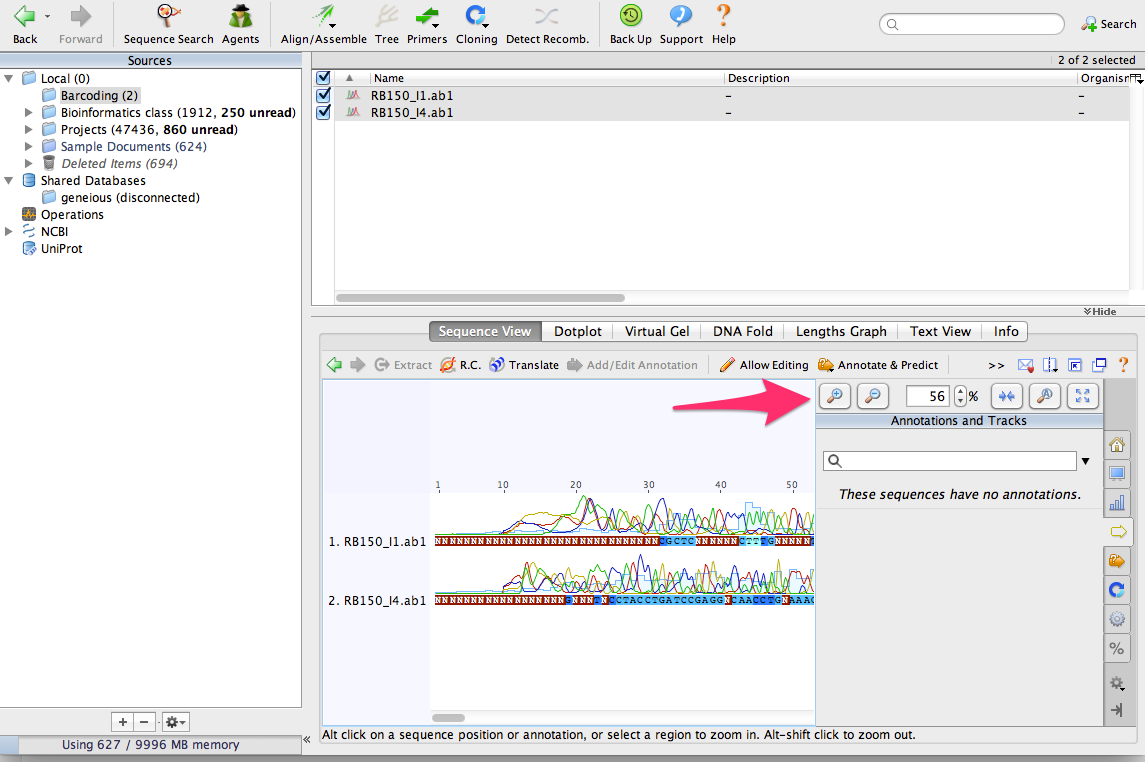
You can see that that there are some sequencing errors, especially at the beginning and end of the sequences. By aligning them together, we’ll be able to correct some of those errors.
Make sure that both sequences are selected. Then, click on the button Align/Assemble. From the menu, select De Novo Assemble…
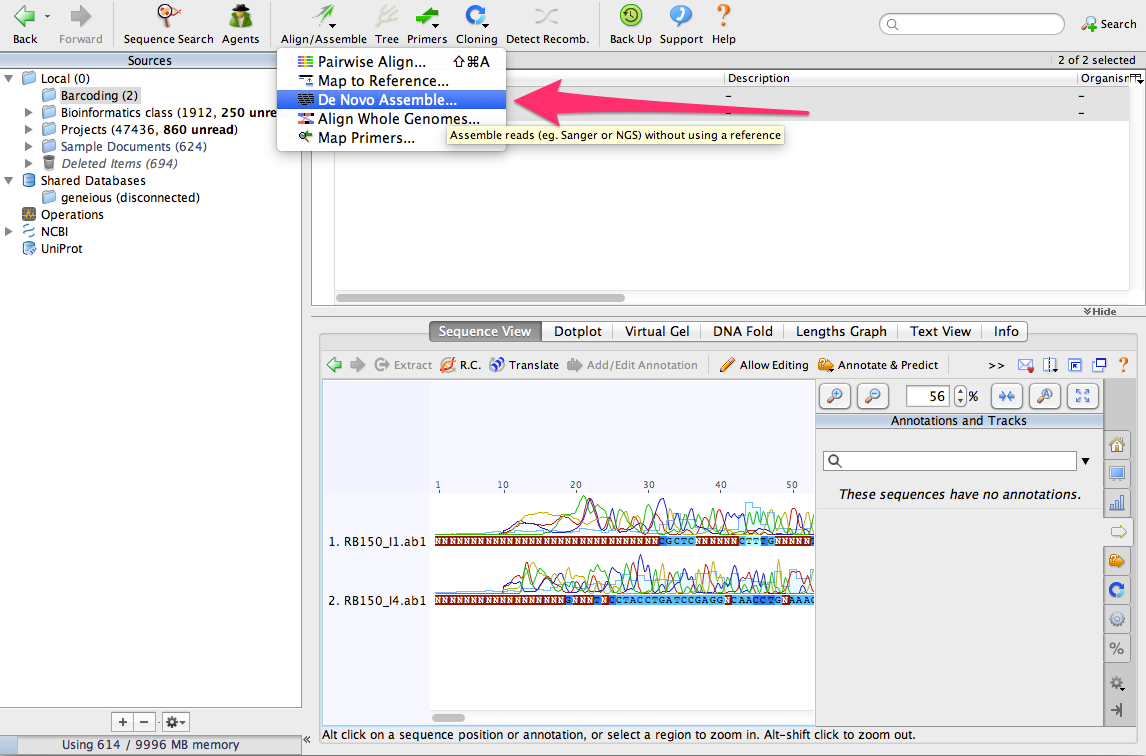
A window will open a new window where you can configure and then run the assembly of the sequences:
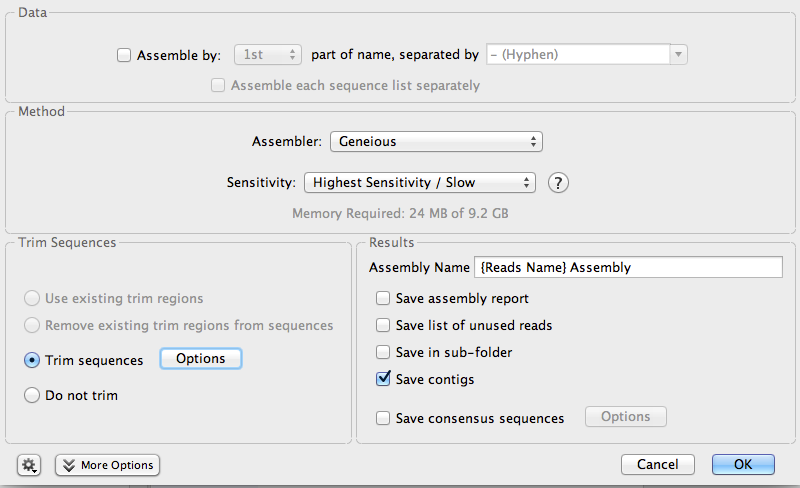
Make sure your settings are the same as in the image.
Now, click on the button Options, next to Trim sequences:
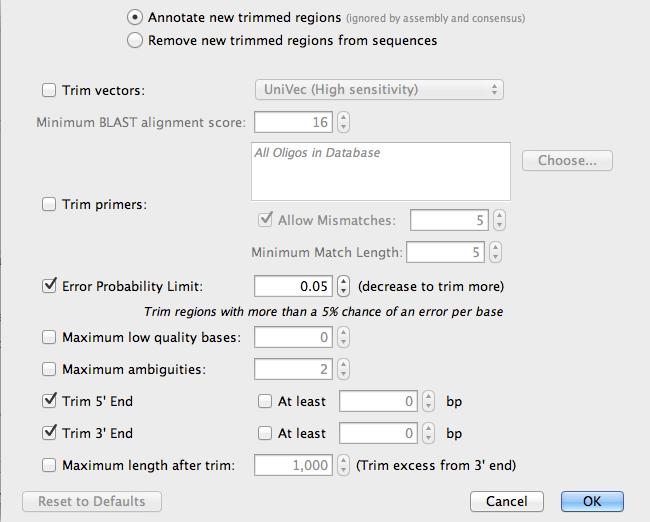
Geneious will automatically detect the low quality bases as the ends of the sequences and mark them (Annotate them) so they will be ignored during the assembly process. Click OK.
Click OK on the De novo Assemble window. Geneious will align the sequences together and make a contig. You can see that Geneious made a reverse-complement of one of the sequences:
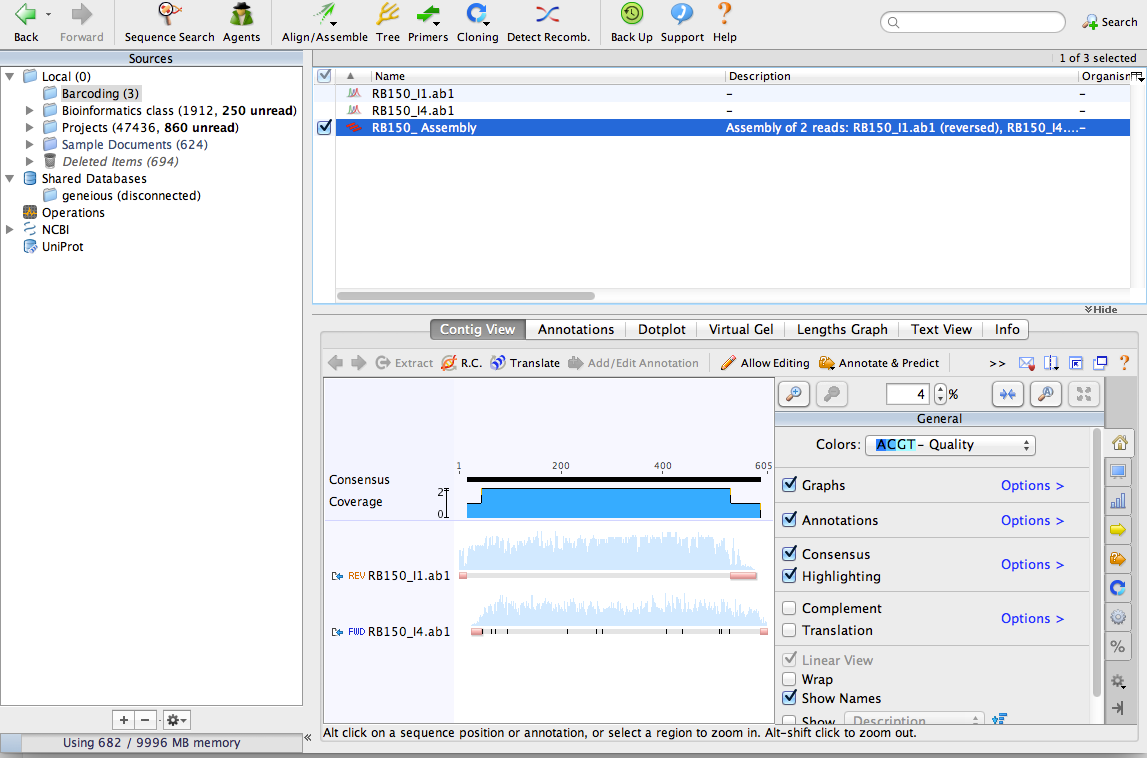
Zoom in until you can see the DNA sequence. You can see the consensus sequence at the top of the contig. You can also see that one of the sequences contains a few N characters. These represent ambiguous bases that could not be detected by the sequencing process. By aligning the two sequences together, we can use the complementary sequence to correct these errors.
Now lets extract a consensus sequence from the contig. Click on the consensus and select only the part where the two sequences overlap. This is the highest quality part of the contig and the part we’ll use as our ‘barcode.’

Click on the button Extract. Geneious will create a new DNA sequence.
Part 2: BLAST searching.
Now we’re going to search GenBank, and find sequences similar to the consensus sequence.
Make sure the consensus sequence is selected, then click the Sequence Search button. A new window will open:
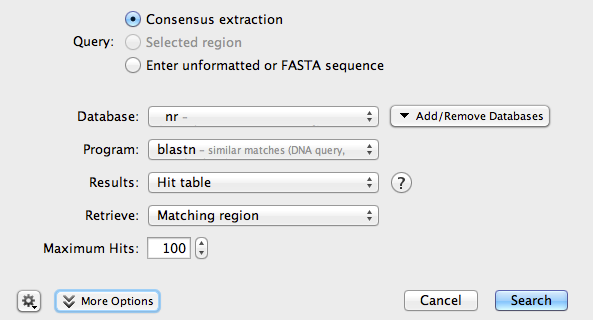
Make sure your search is configured as shown in the picture. Then, click Search.
The search will take a few minutes. When its done, your screen will look like this:
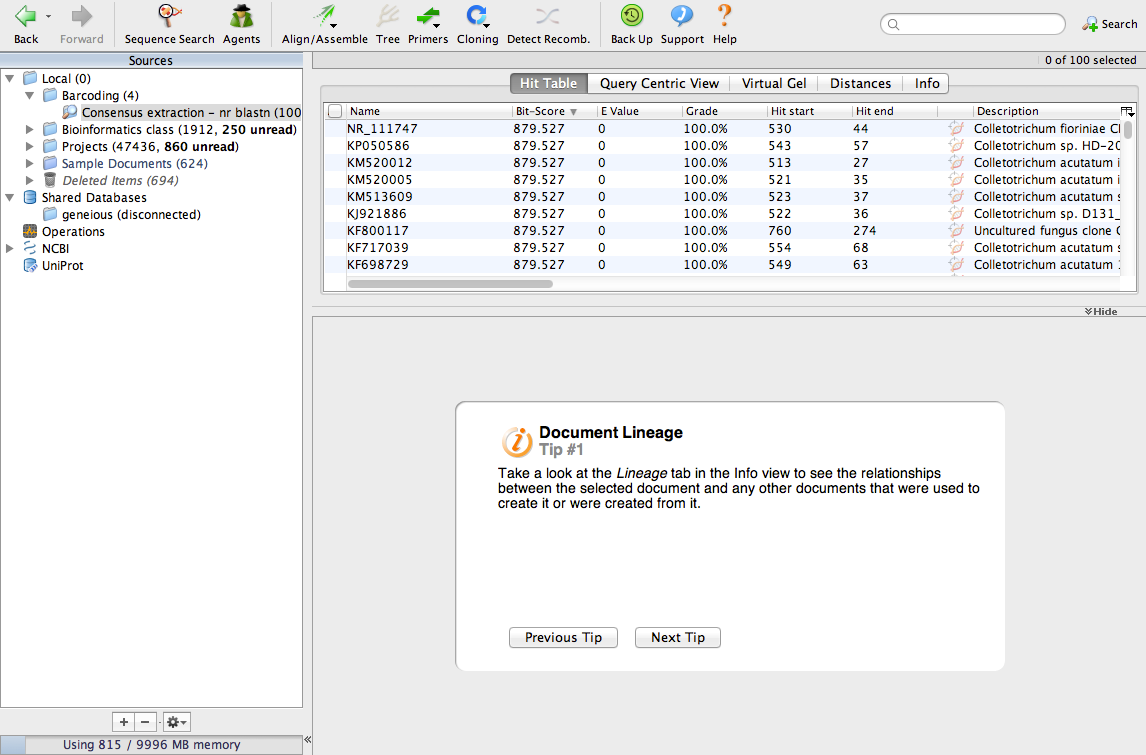
We have identified the 100 most similar sequences to ours. These are usually called BLAST hits. Scroll the table to the right and look for a column called Query Coverage.
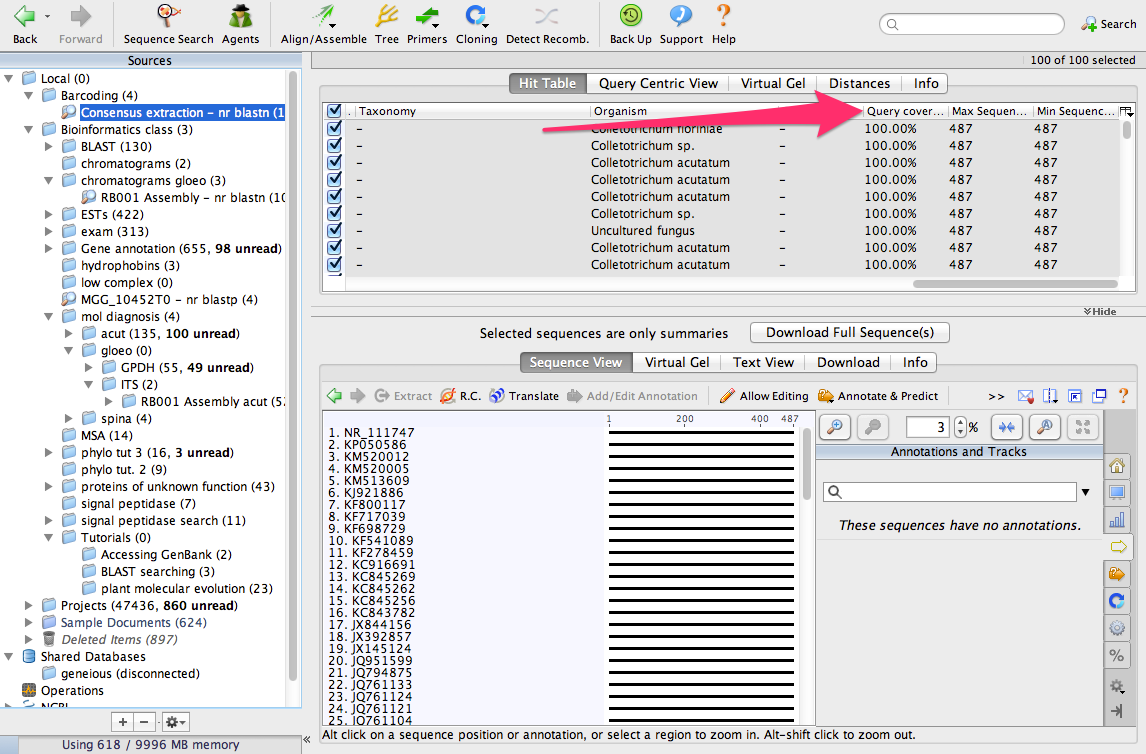
You will see that nearly all of the sequences have a query coverage of 100%. That means that the consensus sequence aligns over its full length to these sequences from GenBank.
Now, look for a column called % Pairwise Identity. You can see that most of the BLAST hits are 100% identical to the consensus sequence.
Find the column called Organism. Most of the BLAST hits are from Colletotrichum acutatum and Colletotrichum fiorinae. Our sequence is 100% identical to sequences from C. acutatum and C. fiorinae so our fungus must be one of those. But, which one is it? If you review the literature, you will find that the species C. acutatum was recently divided into several new species, including C. fiorinae. A lot of the sequences in GenBank have not been updated to reflect this change in taxonomy. From this analysis we can conclude that our fungus is C. fiorinae.
Also notice that among the hits are sequences labeled C. gloeosporioides and Colletotrichum sp. Keep in mind that GenBank is a sequence archive and that they don’t validate the species of every sequence in the database. Consequently, mis-identified sequences can be found in GenBank. There are efforts to create curated databases of ITS sequences specifically for fungal DNA barcoding. Two of these are UNITE and www.fungalbarcoding.org