Discovery Environment is an online platform for running bioinformatics applications and analyzing data. It’s provided for free by CyVerse (previously known as iPlant Collaborative – www.cyverse.org), a project by the National Science Foundation, USA to develop computing infrastructure for life science research. In this tutorial I’ll show you how to sign up for an account and start using Discovery Environment.
Before you can use Discovery Environment (DE), you need to sign up for a CyVerse account. Follow this link to sign up:
https://user.cyverse.org/register/
You will need to check your email to complete the registration with CyVerse.
Now you can go to the CyVerse homepage and log in to DE. Go to http://www.iplantcollaborative.org and look for a link to Discovery Environment. This takes you to a page with information about DE. They’ve really hidden the link to log in, though. Scroll down and look for a link called LAUNCH THE DE. click on it and log in with the user ID and password that you created earlier.
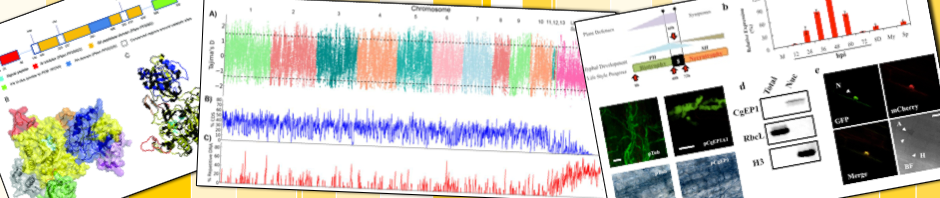
After you log in, the screen should look like this:

Click on the user account icon (1) and then select Introduction from the menu (2). Follow the introduction to learn about the different parts of DE.
Now, let’s use DE to make a multiple sequence alignment. Download this file to your computer: sequences.fasta To download it, click on the link with the right mouse button, then select Download from the menu that appears. The file name might change to sequences.fasta.txt after you download it. Thats OK.
That file contains a few protein sequences in FASTA format. Let’s upload it to DE and make a multiple sequence alignment.
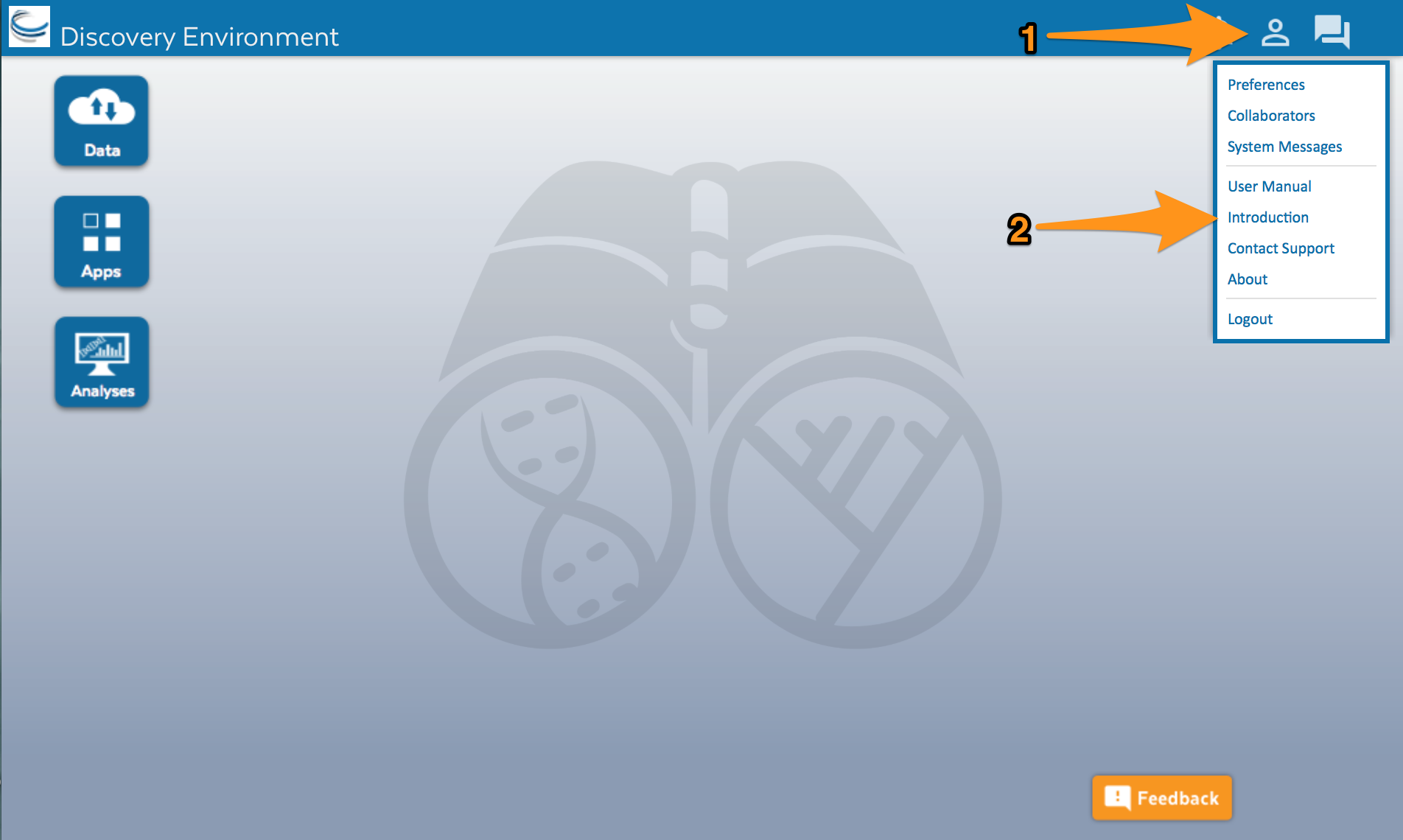
In DE, click on the ‘Data’ button that you see on the left side of the screen. A window will open and you will see several folders on the left side. The first folder has your CyVerse user name. Click on it.
As you can see, I already have some files and folders. Yours may be empty. Now lets upload the sequences.fasta file.
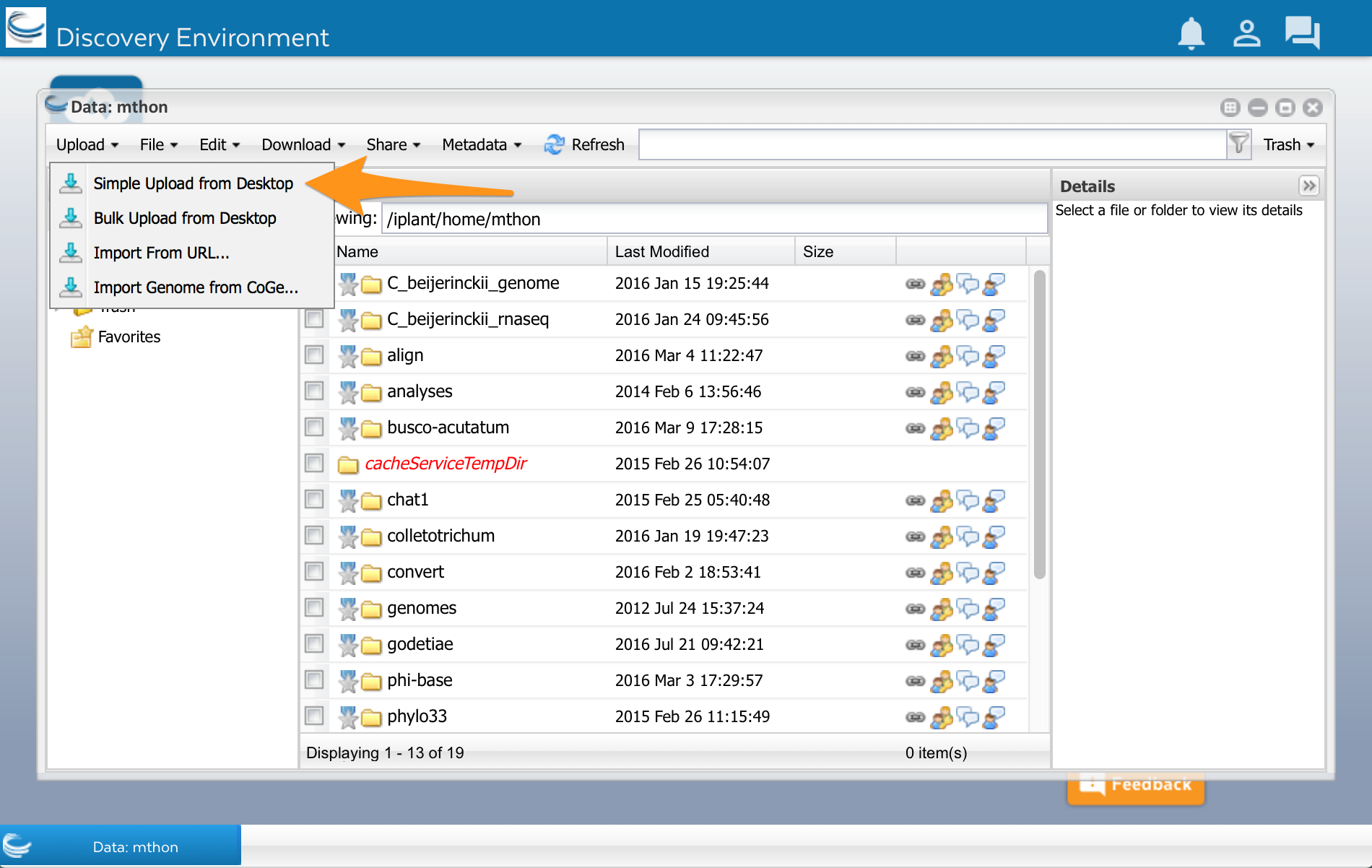
At the top of the Data window, select ‘Upload’ and then ‘Simple Upload from Desktop:
A new window opens. Select Browse, and then select your sequences.fasta file:
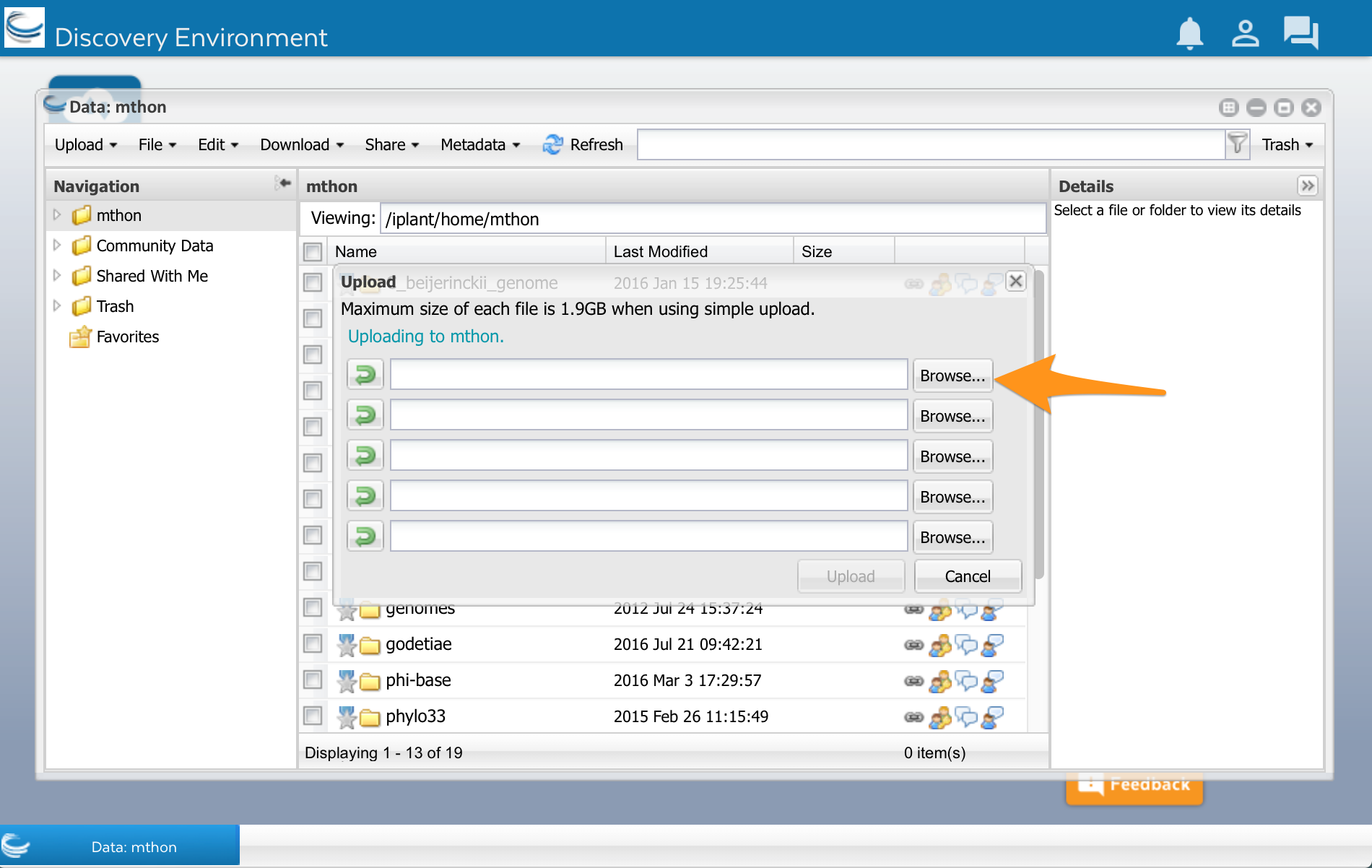
After the file has finished uploading, click the Refresh button. You should see your file appear in DE.
Click on the file name to open it. You can view and edit your sequences in the window.
Now we need to find an app to align the sequences. Close all of the open windows and click on the Apps button. Here you can see a list of apps for analyzing data organized into categories. Let’s search for an app to make a multiple sequence alignment.
Type ‘Muscle’ into the search field at the top of the window (1). Several Muscle apps will appear in the search results:
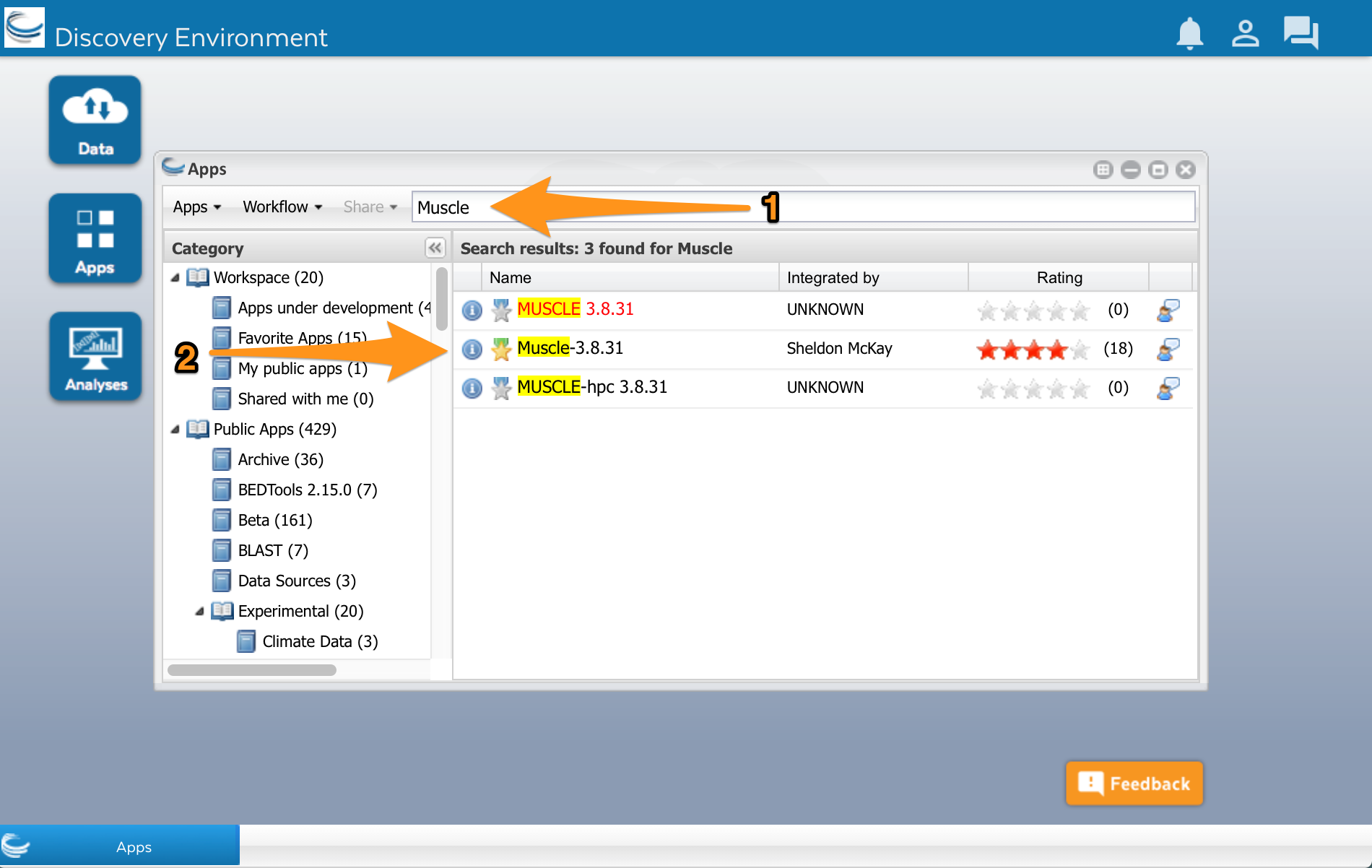
We’ll use the Muscle app developed by Sheldon McKay (2). Click on the app’s name to use it. A new window will open. Apps are usually divided into two areas where we can configure the app and select the input files before running it. The muscle app has three areas: Analysis name, Select input data, and Sequence Type.
There is a row called ‘Select input data.’ Open that up by clicking on the little triangle to the right:
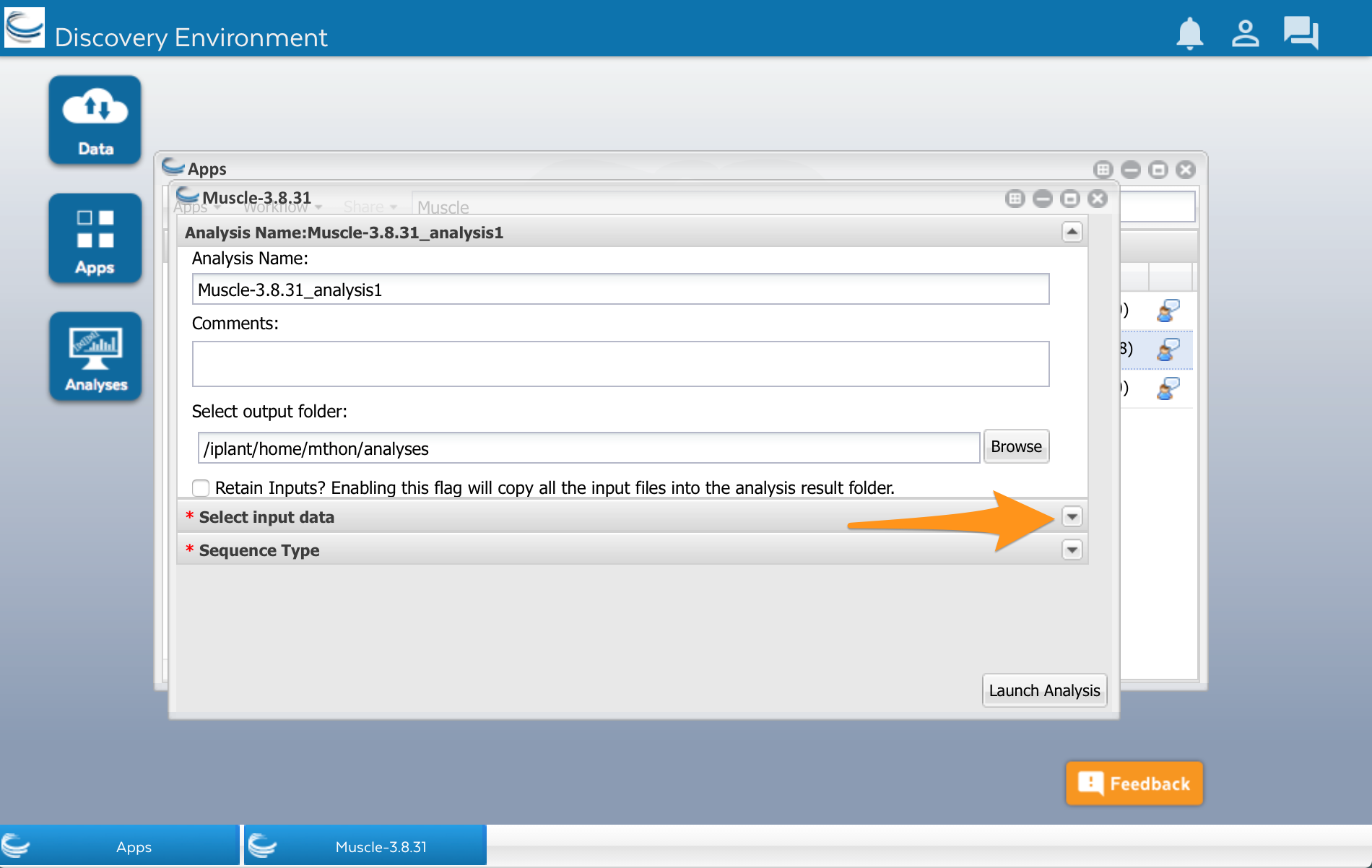
After clicking that little triangle, you should see a field that will let you select the file of sequences that you want to align:
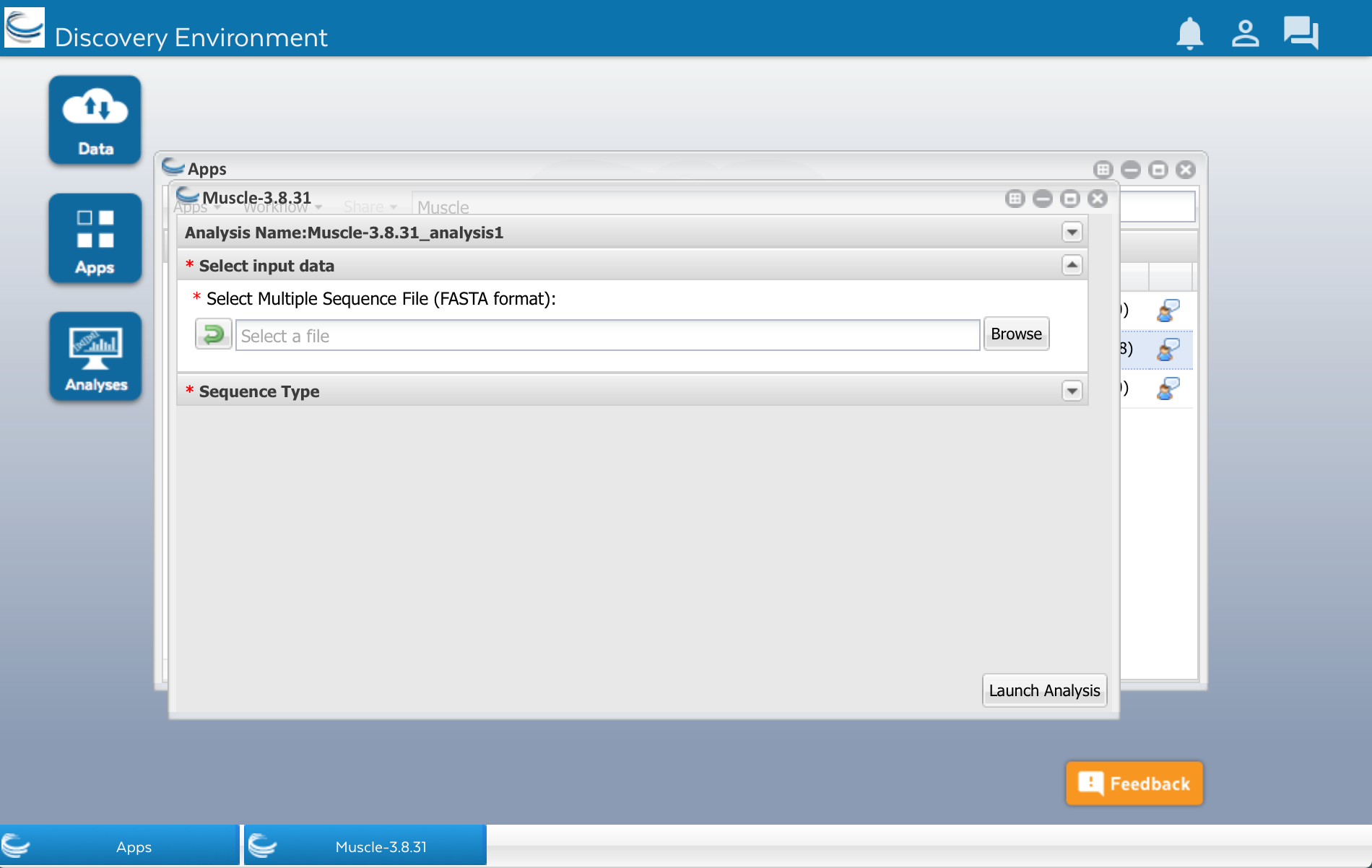
Click the Browse button. Find your sequences.fasta file in the list and select it.
Now you need to select the type of sequences in your file. Click on the arrow next to ‘Sequence Type’ to open it. Select ‘Auto’ from the list. This tells the app to automatically determine whether your sequences are DNA or protein.
Now you’re ready to launch the app. Click the Launch Analysis button.
Click on the ‘Analyses’ button. A new window will open showing you a list of apps that you have run or are currently running. Analyses will have different statuses. Submitted, Running and Completed are the most common. Keep in mind that your analysis will run on a large computer cluster that is shared with researchers all over the world. When you click the Launch analysis button of an app, your analysis is added to a queue. The server will wait until a computer (part of the computer cluster) is available, and then run your analysis.
Wait a few minutes, then click on the Refresh button to update the status. It should change to Running or Completed.
Now lets look at the results. Open the data window using the Data button. Click on the little triangles next to the folders to open the analyses folder. You should have a folder called ‘Muscle.’ Select it, and you will see a list of files. You can see your multiple sequence alignment in four different formats. You can also see another folder called ‘logs.’ This folder contains files with details about how the analysis was done. You normally don’t need these files, but they might be useful if an analysis fails or you get unexpected results.
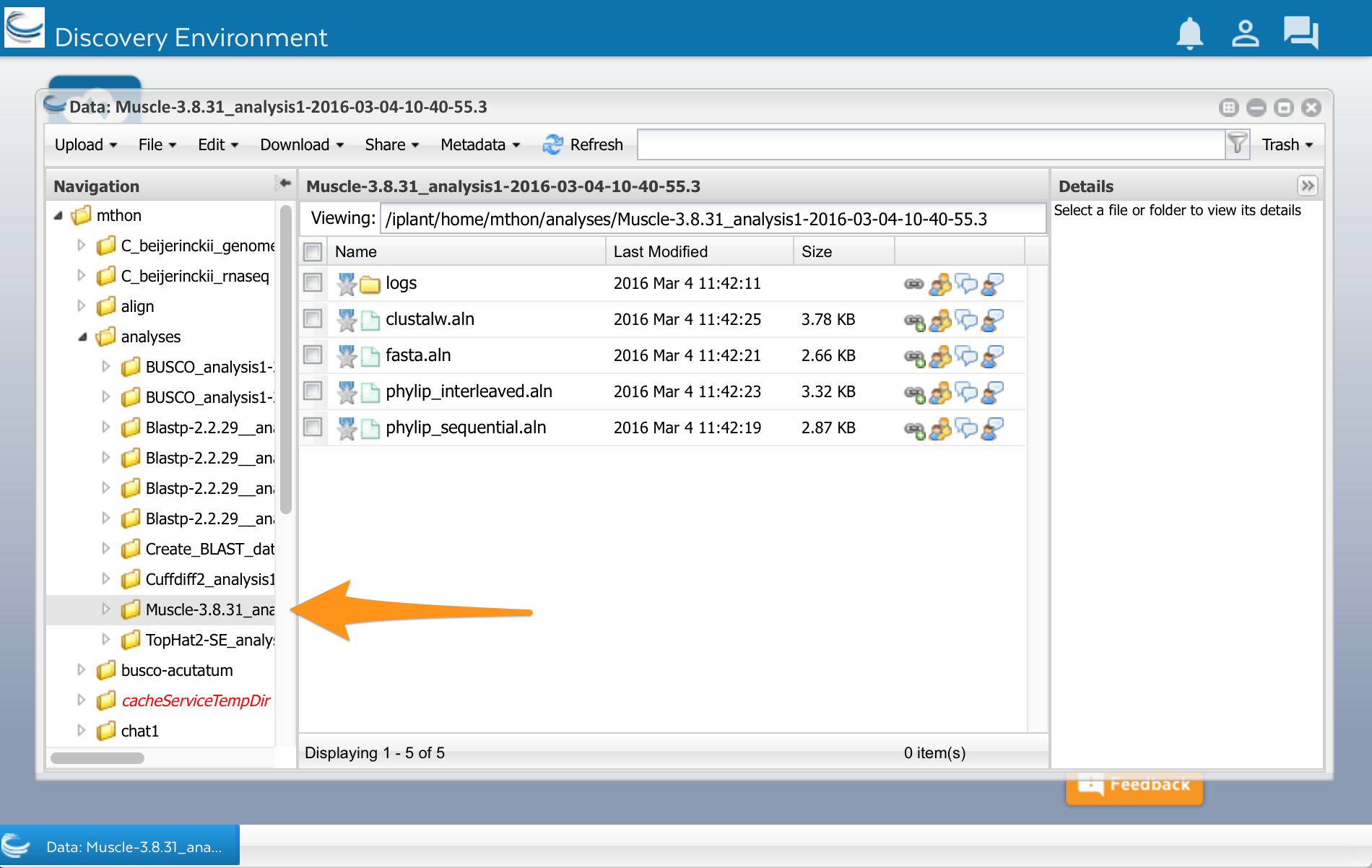
You can click on the files to open them. You can also use them in other analyses, for example, to make a phylogenetic tree.
With DE, you can do a lot more that align sequences. It’s real power comes with its ability to process genome-scale data sets, including RNA-Seq analysis, SNP detection, metagenomics, and more.
This tutorial was based on the Quick Start tutorial on the CyVerse web site.